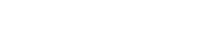


Nature:巨噬细胞ETS2信号在多种炎症性疾病重要作用
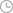 2024-06-17
2024-06-17自身免疫和炎症性疾病对人类健康构成重大威胁,现有治疗方法效果有限,迫切需要了解疾病机制。遗传学提供了研究疾病机制的独特机会,但大多数风险变异体并不位于编码DNA中,而是在不太保守的非编码基因组区域。
大多数非编码变异体被认为影响基因调控,但识别因果基因(可能位于数百万个碱基之外)和因果细胞类型(可能在特定条件下仅表达相关基因)的困难阻碍了确定疾病机制的努力。例如,尽管基因组关联研究(GWASs)已经识别了超过240个IBD风险位点,包括几个可能的药物靶点,但只有少数几个机制上得到了解决。
2024年6月5日,Francis Crick研究所J. C. Lee通讯在《Nature》发表“A disease-associated gene desert directs macrophage inflammation through ETS2”的研究论文。这项研究探讨了某些遗传变异如何导致多种疾病,特别是在21号染色体上的一个非编码区域(chr21q22)。


染色体21q22上的一个间区区域(chr21q22)与五种炎症性疾病有关,这个区域因为缺乏编码基因,最初被认为是“基因沙漠”,但常常包含基因组关联研究(GWASs)的显著位点。
为了探索共同的疾病机制,研究者们进行了共定位分析,并确认了每种疾病的遗传基础是相同的,这意味着存在一个或多个共同的因果变异,以及一个共享的分子效应。
由于这些异质性疾病都是免疫介导的,研究者们推断这个位点必须包含一个在免疫细胞中发挥作用的远端增强子。通过分析H3K27ac染色质免疫沉淀测序(ChIP-seq)数据,研究者们在这个位点鉴定了一个单核细胞/巨噬细胞特异性的增强子。
研究者们接下来试图确定这个增强子调控的基因。虽然相关的位点没有编码基因,但之前的研究已经突出了几个附近的候选基因,包括PSMG1、BRWD1和ETS2。利用人类单核细胞的启动子捕获Hi-C和表达定量位点(eQTL)数据,研究者们发现疾病相关的位点与ETS2的启动子有物理交互,并且风险等位基因与ETS2表达升高有关。
通过CRISPR-Cas9技术删除chr21q22增强子区域后,研究者们发现单核细胞在受到炎症配体刺激时,ETS2的上调表达显著减少,证实了这一多效性位点包含一个远端的ETS2增强子。
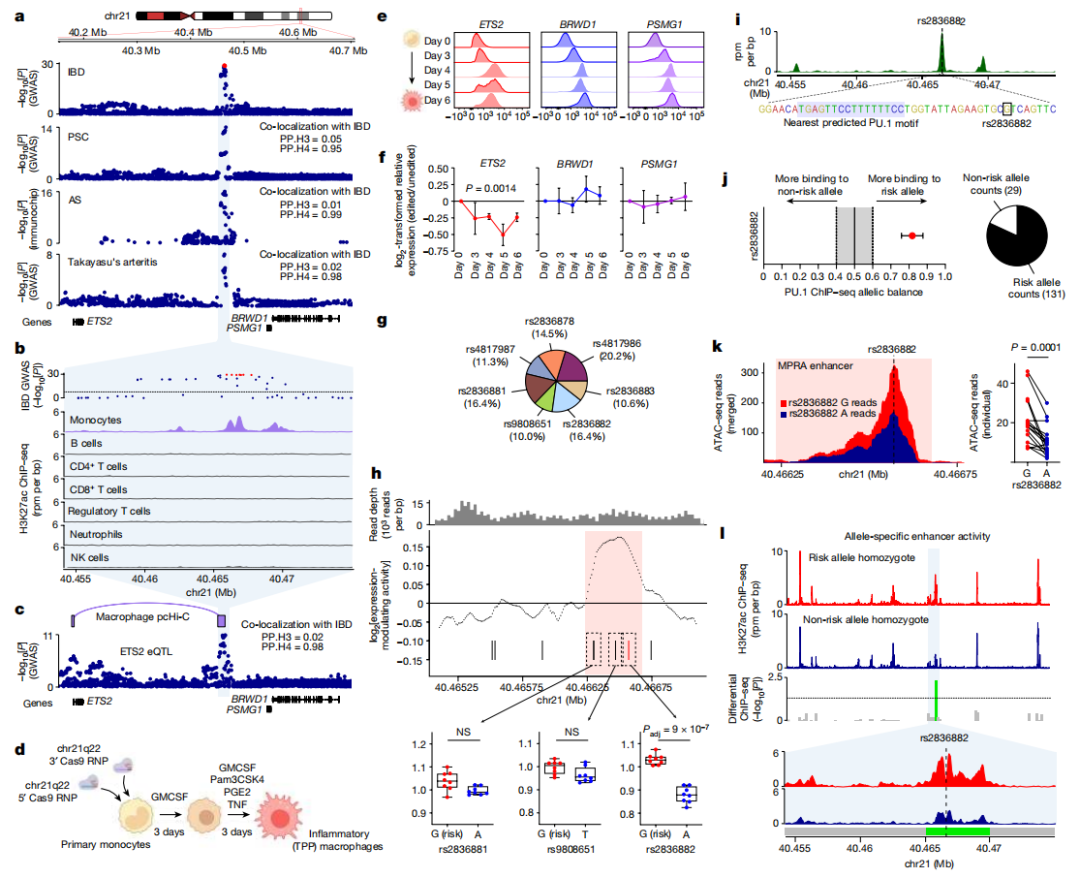
为了确定因果变异,研究者们大型IBD GWAS3中进行了统计精细定位,但遗憾的是,由于候选单核苷酸多态性(SNPs)之间的连锁不平衡较高,这并没有解决关联问题。
于是,研究者们采用了功能性方法,首先描绘了该位点上的活跃增强子,然后是否有候选 SNP 可能会改变增强子的活性。这种方法,即大规模平行报告分析(MPRA),可以同时测试成千上万的短DNA序列的增强子活性。
通过滑动窗口分析,研究者们确定了一个单一的442 bp的增强子活性焦点,包含三个(共七个)候选SNPs。其中两个多态性在转录上是惰性的,但第三个(rs2836882)对转录有最强的调节作用,风险等位基因(G)增加了转录,与ETS2 eQTL一致。这个SNP位于每个共定位分子特征的可信集合中,并且位于巨噬细胞PU.1 ChIP-seq峰内。通过分析来自杂合子巨噬细胞的PU.1 ChIP-seq数据,研究者们检测到了与风险等位基因的强烈特异性结合。
为了测试内源位点上等位基因特异性的增强子活性,研究者们对来自rs2836882主要和次要等位基因纯合子的炎症巨噬细胞进行了H3K27ac ChIP-seq分析。虽然大多数chr21q22增强子峰在这些捐赠者之间是相似的,但覆盖rs2836882的增强子活性在主要(风险)等位基因纯合子中显著更强,该位点活性大约增加了2.5倍。
综上,这些数据揭示了一个机制,即chr21q22上的假设因果变异通过其在原代巨噬细胞中的功能性效应,促进先锋因子的结合,增强染色质可及性,并增加远端ETS2增强子的活性。
ETS2是ETS家族转录因子和原癌基因,但其在人类巨噬细胞中的具体作用尚不明确。
为了明确ETS2在人类巨噬细胞中的作用,并确定ETS2表达失调如何导致疾病,研究者们首先使用一种基于CRISPR-Cas9的敲除功能方法。为了控制脱靶效应,设计了两种靶向不同ETS2外显子的gRNA,分别进行了验证,并单独整合到Cas9核糖核蛋白中,用于转染原代单核细胞。
它们分别在约 90% 和 79% 的细胞中产生了靶向编辑,并有效降低了ETS2的表达。细胞存活率和巨噬细胞标志物的表达没有受到影响,这表明ETS2对于巨噬细胞的存活或分化不是必需的。相比之下,在ETS2破坏后,包括IL-6、IL-8和IL-1β在内的促炎性细胞因子产生明显减少,而抗炎性细胞因子IL-10则影响较小。
研究者们接下来调查了ETS2破坏是否会影响巨噬细胞的其他功能。通过流式细胞术检测,他们发现ETS2破坏后,吞噬作用同样受到损害。
他们还检测了细胞外活性氧(ROS)的产生,破坏ETS2极大地降低了巨噬细胞的氧化爆发。
为了理解这些效应的分子基础,研究者们对来自多个供体的经 ETS2 编辑和未经编辑的炎症巨噬细胞进行了 RNA 测序(RNA-seq)。ETS2的破坏导致了广泛的转录变化,包括许多炎症基因的表达减少。这些基因包括细胞因子(如TNFSF10/TRAIL、TNFSF13、IL1A和IL1B)、趋化因子(如CXCL3、CXCL5、CCL2和CCL5)、分泌效应分子(如S100A8、S100A9、MMP14和MMP9)、细胞表面受体(如FCGR2A、FCGR2C和TREM1)、模式识别受体(如TLR2、TLR6和NOD2)和信号分子(如MAP2K、GPR84和NLRP3)。
为了更好地描述受影响的通路,研究者们使用基因本体(GO)生物通路数据集进行了基因组富集分析(fGSEA)。这证实了功能缺陷,其中负向富集最多的通路(通过ETS2破坏而下调)与巨噬细胞激活、炎症细胞因子产生、吞噬作用和ROS产生有关。ETS2破坏后上调的基因较少,但有氧呼吸和氧化磷酸化(OXPHOS)出现了正富集——这些代谢过程与抗炎表型相关。
总的来说,这些数据揭示了ETS2在巨噬细胞炎症反应中的关键作用,这可能解释了为什么ETS2 表达失调会增加疾病风险。
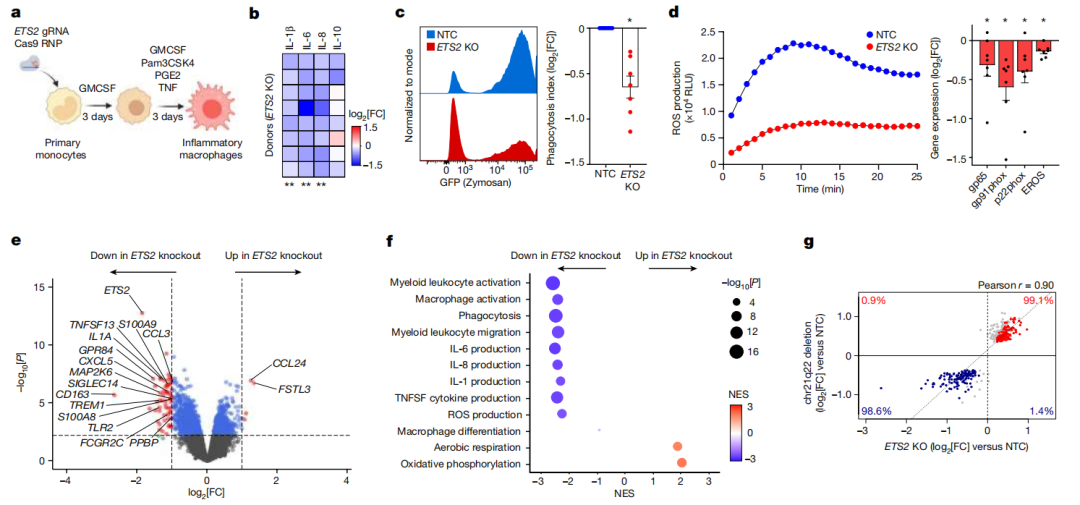
接下来,研究者们研究了增加ETS2表达的效果。为了实现这一点,他们优化了一种方法,通过转染体外转录的ETS2 mRNA,以最小化免疫原性,从而在原代巨噬细胞中控制目标基因的过表达。在转染过程中,细胞暴露于低剂量的脂多糖,以启动可能被放大的低度炎症反应。
他们发现,过表达ETS2会增加促炎性细胞因子的分泌,而IL-10再次受到影响较小。
为了更好地描述这种反应,研究者们进行了RNA测序,并重新审视了需要ETS2的炎症通路。值得注意的是,所有这些通路——包括巨噬细胞激活、细胞因子产生、ROS产生、吞噬作用和迁移——都以剂量依赖的方式被ETS2过表达诱导,当转染更多的ETS2 mRNA时,每个途径的富集程度都更大。
这表明ETS2在人类巨噬细胞中既是炎症反应的必要条件,也是充分条件,它是效应器功能的核心调节因子,其失调与疾病直接相关。
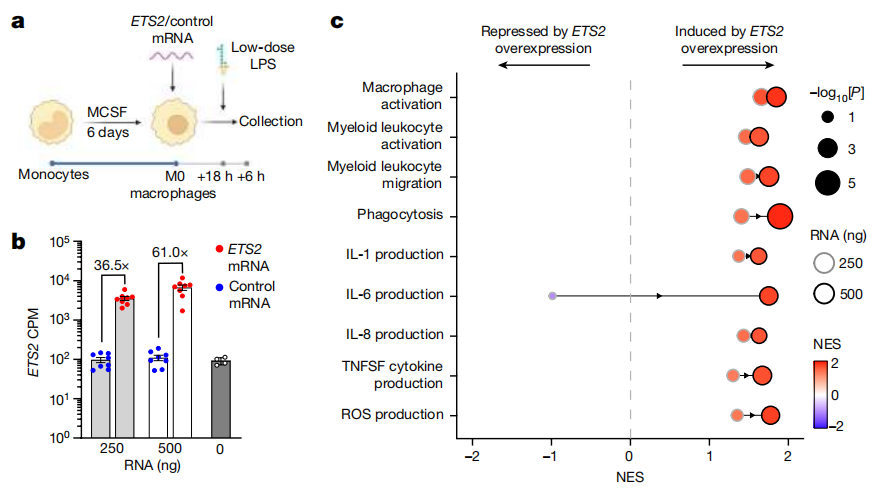
为了测试ETS2是否参与疾病中巨噬细胞的表型,研究者们比较了在静息巨噬细胞中过表达ETS2的效果,以及来自克罗恩病肠道巨噬细胞的单细胞RNA测序(scRNA-seq)特征。ETS2过表达诱导了一个与疾病巨噬细胞非常相似的转录状态,包括几个治疗靶点的核心(前沿)富集。来自其他 chr21q22 相关疾病的骨髓特征基因也出现了类似的富集,在较小程度上,来自活动性细菌感染的骨髓特征基因也出现了类似的富集,但来自流感和肿瘤巨噬细胞的特征基因却没有出现类似的富集,这表明ETS2并不是简单地诱导通用的激活。
鉴于ETS2在炎症性巨噬细胞中的核心作用,以及这些细胞在疾病中的重要性,研究者们假设其他遗传关联也会涉及这个途径。
GWAS的主要目标是识别疾病途径,但由于确定因果基因和变异体的数量有限,这证明是一项具有挑战性的任务。为了确定巨噬细胞ETS2途径是否富含疾病遗传学,研究者们专注于IBD,因为它比其他任何与 chr21q22 相关的疾病拥有更多的GWAS显著位点。
令人鼓舞的是,之前在肠道粘膜中发现了33个与IBD相关的基因网络富含预测的ETS2基序。
通过研究在 ETS2-修饰的巨噬细胞中一致下调的基因,研究者们发现了超过20个与IBD风险相关的基因,其中包括许多被认为在其各自位点具有因果作用的基因。
为了更好地测试IBD GWAS显著位点在ETS2介导的炎症中的富集程度,并将其与已知的疾病通路进行比较,研究者们使用了SNPsea,这是一种识别疾病位点影响途径的方法。总共测试了241个IBD位点,涉及7,658个GO生物途径和2个重叠的ETS2调控基因列表(要么是由ETS2破坏下调,要么是由ETS2过表达上调)。
值得注意的是,与IBD相关的SNPs在巨噬细胞ETS2途径中的富集程度比许多IBD途径都要显著,没有任何一个无效的SNP集在这两个ETS2调控基因列表中更富集。与原发性硬化性胆管炎、强直性脊柱炎和多发性大动脉炎相关的SNPs也富集在ETS2目标基因中。
总的来说,这表明巨噬细胞ETS2信号在多种炎症性疾病中起着核心作用。
接下来,研究者们调查了ETS2如何控制如此多样的巨噬细胞功能。
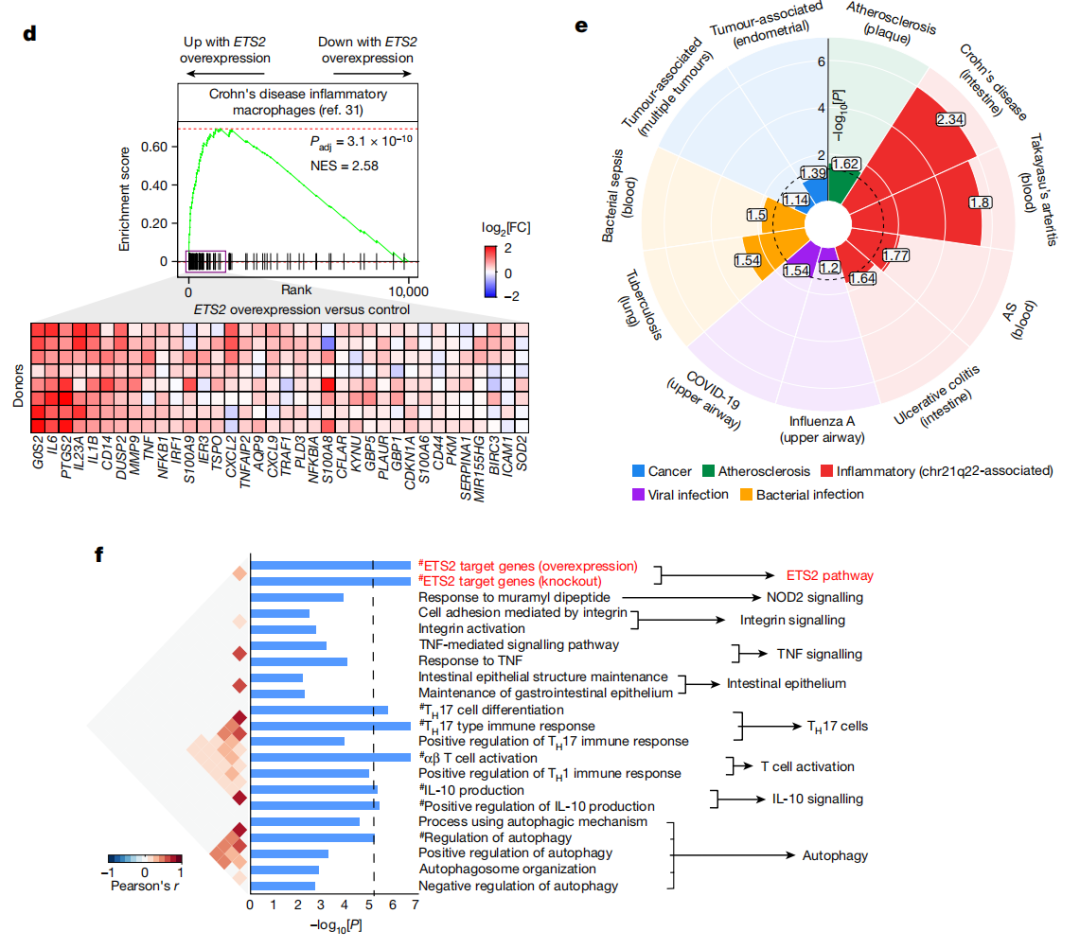
研究ETS2的生物学具有挑战性,因为没有ChIP级抗体,这直接阻止了对其转录靶点的直接识别。因此,他们首先使用一种关联性方法来识别在67种人类巨噬细胞激活条件下与ETS2共表达的基因。这揭示了PFKFB3——编码糖酵解的限速酶——与ETS2最强烈共表达,HIF1A也高度共表达。
这些基因共同促进了一种“糖酵解开关”,这是髓系炎症反应所必需的,因此,研究者们推测ETS2可能通过代谢重编程控制炎症,OXPHOS基因与ETS2负相关的,以及在ETS2破坏后上调支持了这种可能性。
为了评估破坏ETS2的代谢后果,研究者们使用气相色谱耦合质谱(GC-MS)量化了编辑过和未编辑过的TPP巨噬细胞中13C-葡萄糖的标记摄入量。ETS2破坏后,检测到标记的葡萄糖代谢物和总葡萄糖代谢物普遍适度减少。
为了确定代谢变化是否解释了ETS2介导的炎症效应,研究者们用roxadustat(一种促进糖酵解的HIF1α稳定剂)处理ETS2编辑的巨噬细胞,这影响了糖酵解和OXPHOS基因,但没有挽救ETS2破坏的影响,无论是转录上还是功能上。
因此,尽管破坏ETS2会损害巨噬细胞的糖代谢,但这并不能完全解释炎症上的差异。
于是,研究者们重新审视了是否可以直接识别ETS2的靶基因。由于ChIP-seq涉及可能改变蛋白质表位和阻止抗体结合的步骤(如固定),他们测试了是否有抗ETS2抗体可用于靶标下的裂解和使用核酸酶释放(CUT&RUN),这不需要这些步骤。
一种抗体识别了多个显著富集的基因组区域(峰),其中6,560个峰在两个生物重复中被重复检测到,并且质量指标可接受。这些峰主要位于活跃的调控区域(90%在启动子或增强子中),并且高度富集富集典型的ETS2基序和已知的ETS2相互作用者基序,包括FOS、JUN和NF-κB。
此外,研究者们还在chr21q22增强子上检测到了ETS2结合。
这些数据表明,ETS2是单核细胞和巨噬细胞炎症反应的核心调节因子,它能够指导多方面的效应程序,并创造一个有利于炎症的代谢环境。
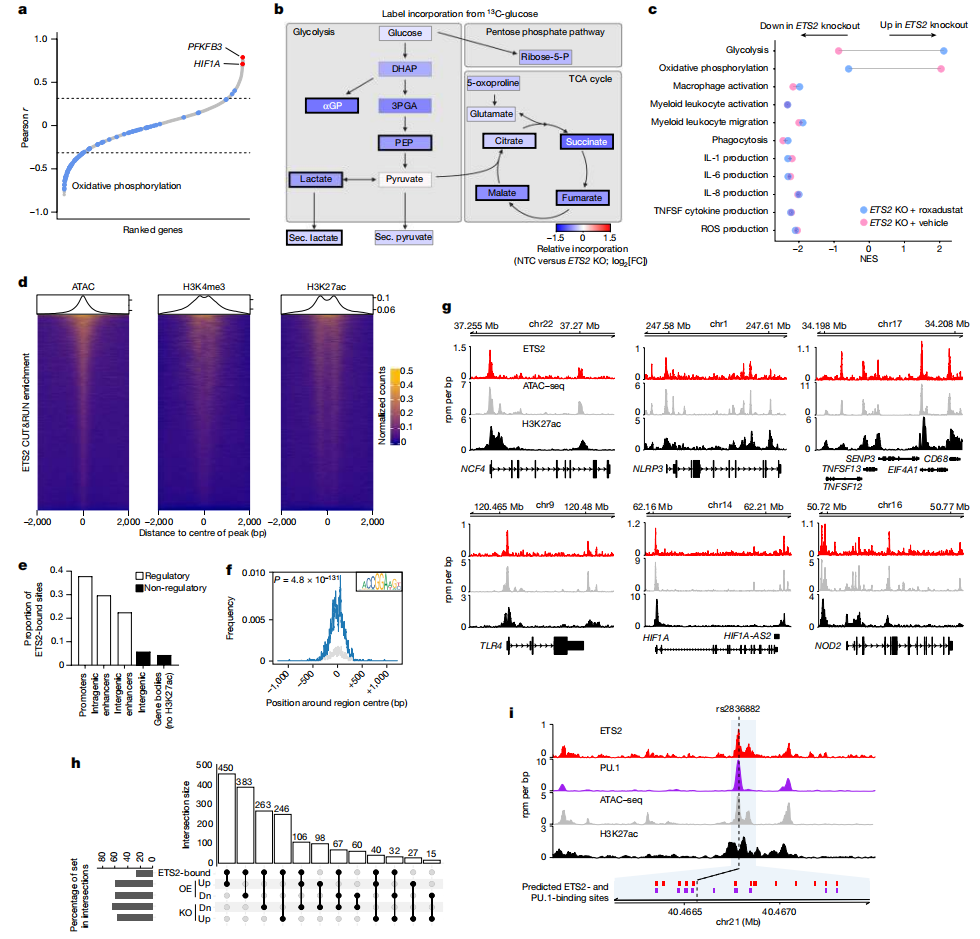
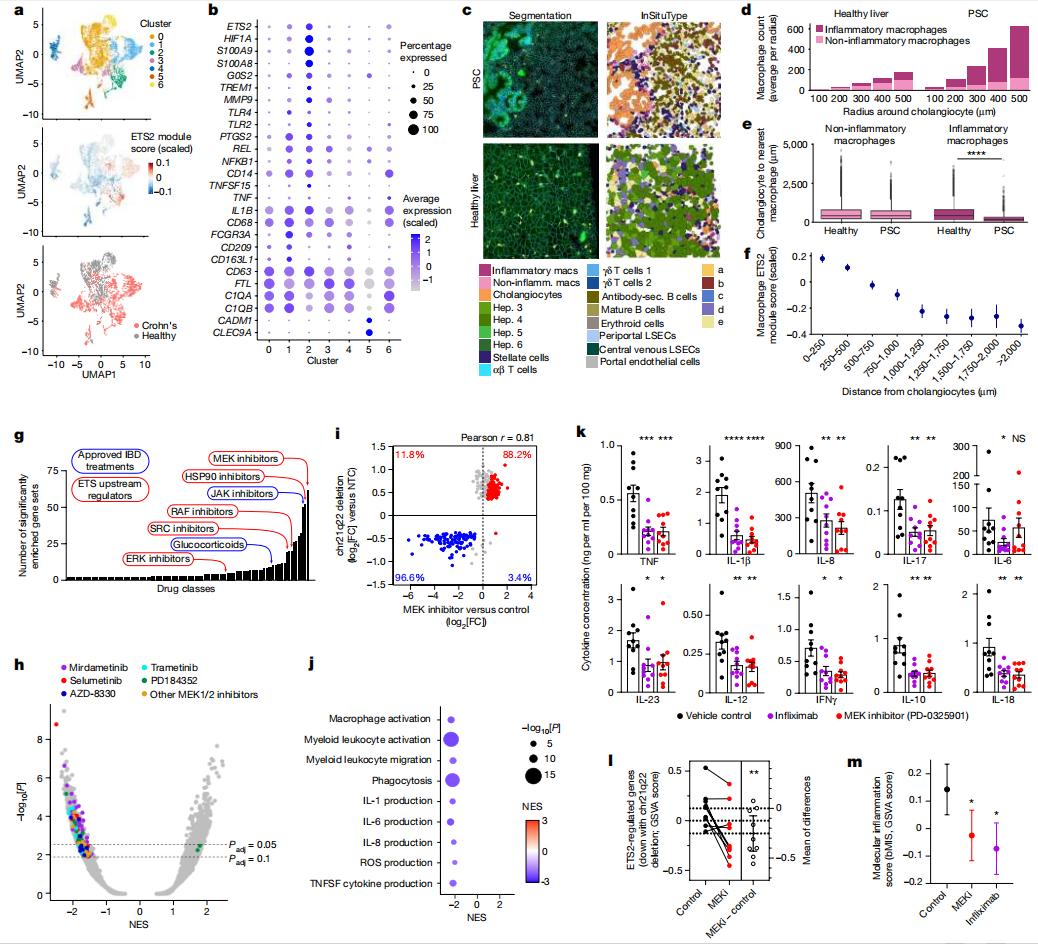
为了评估ETS2如何影响疾病组织中巨噬细胞的异质性,以及是否可以作为治疗目标,研究者们检查了来自克罗恩病患者和健康对照个体的肠道单细胞RNA测序数据。在髓系细胞中,研究者们发现了7个集群。疾病中炎症性巨噬细胞(表达CD209、CCL4、IL1B和FCGR3A的集群1)和炎症性单核细胞(表达S100A8/A9、TREM1、CD14和MMP9的集群2)被扩大,并ETS2及其调控基因的表达水平高于其他集群,包括组织驻留巨噬细胞(表达C1QA、C1QB、FTL和CD63的集群0)和常规树突状细胞(表达CLEC9A、CADM1和XCR1的集群5)。
使用空间转录组学技术,研究者们在PSC肝脏组织中观察到类似的炎症性巨噬细胞增加,这些细胞与胆管上皮细胞紧密接触。值得注意的是,巨噬细胞与胆管上皮细胞越接近,ETS2调控基因的表达越高。
事实上,使用批量RNA测序数据,研究者们发现ETS2的转录足迹在多个chr21q22相关疾病的受影响组织中可检测到。
接下来,研究者们研究了是否可以通过药物靶向这一途径。目前还没有特定的ETS2抑制剂,而且结构分析表明没有明显的变构抑制机制。因此,他们使用NIHLINCS数据库来识别可能调节ETS2活性的药物。
使用基因集富集分析,研究者们发现906个特征模拟了破坏ETS2的效果,包括几种批准的IBD疗法。最大的药物类别是一类药物是MEK抑制剂。这一结果并非单一化合物所致,而是多种MEK1/2抑制剂下调ETS2靶基因的类药物效应。
为了测试MEK抑制是否能减轻ETS2驱动的人类巨噬细胞炎症,研究者们用用选择性非ATP竞争性MEK抑制剂PD-0325901治疗TPP巨噬细胞,观察到强烈的抗炎作用,其表型与破坏ETS2或chr21q22增强子的效果相同。
为了进一步评估治疗潜力,研究者们分别用MEK抑制剂或阴性或阳性对照培养了活动性、未经治疗的IBD的肠道活检样本。MEK抑制减少了炎症性细胞因子的释放到与英夫利昔单抗(一种广泛用于IBD的抗TNF抗体)相似的水平。此外,ETS2调控基因的表达减少,并且炎症的转录评分有所改善。
这些数据表明,靶向ETS2的上游调节器可以阻断chr21q22相关疾病中的病理炎症,并且可能具有治疗价值。